Since the introduction of several new standards in recent years and the increase in scrutiny of data collection and management, there has been an increase need to qualify and quantitate the use of this data to satisfy quality expectations. The principles described below demonstrate an example of the development and use of a risk based model to manage alarms, alerts and notifications to satisfy both GMP requirements, as well as business and engineering/maintenance needs.
Given recent observations, 483’s and warning letters over alarming (or lack of it) within the manufacturing environment, the development of standards or guidance to engineering and Quality Assurance is becoming an expectation for each company.
GMPs and Alarm Management
Alarm management principles, together with the ISPE guidelines for risk management, provide a good foundation for the implementation of alarm systems in pharmaceutical manufacturing processes. This article provides an overview of the key principles of Alarm Management and how these can be used by the pharmaceutical industry to implement a risk based approach to alarm management to enhance productivity and implement exception reporting. A brief overview of alarm management literature and explanation of the alarm concept are provided with the aim of furthering insight and understanding.
Background
In practice, the accepted definition and primary function for alarms is to warn or alert of an unsafe condition. Alarm Management standards define that a necessary attribute for any alarm is its response procedure . If an alarm does not warrant a response; then, the alarm does not have a purpose, and should be removed from the alarm system. The response procedure defines the purpose and actions required to mitigate the alarm condition. The most critical use of alarms is to warn people of environmental or life threatening danger.
For example, a Building Fire Alarm, an Aircraft Engine Trouble Alarm, and Tornado Alarms. Secondary to safety, alarms are used in process control instrumented systems to indicate unexpected or abnormal process condition. For example, fermentation processes implement process out of tolerance alarms and automated controls of temperature, pressure, oxygen, and carbon dioxide. A third key use for alarms is to indicate equipment malfunctions; e.g. motor fail to start, and network communication failure.
Process and equipment failure alarms are used to warn of process ineffectiveness, to prevent process discrepancies, rework, and product loss. Based on these examples, a comprehensive definition of alarm can be deduced: to warn or alert and provide response methods for 1) unsafe conditions, 2) process out of tolerance conditions, and 3) equipment failures. Other authors identify using alarms to warn for Inefficient Operation.
Several standards have been written about Alarm Management. One of the most influential publications, dating back to 1999 and revised in 2007, is the Engineering Equipment Manufacturers and Users Association (EEMUA) Publication 191, Alarm Systems – A Guide to Design, Management and Procurement. In 2009 ANSI/ISA approved the standard ANSI/ISA S18.2, Management of Alarm Systems for the Process Industries. A great source for effective alarm management is the ASM Effective Alarm Management Practices, 2009. The focus of standards literature is to describe the need for companies to define an alarm management philosophy, and in how to implement an effective alarm management life cycle. The objective of the alarm management philosophy is to document the purpose, scope, and resources necessary for the effective use of alarms. Objectives should include personnel and environmental safety, optimizing process efficiency and ultimately profitability.
The key aspects of alarm management can be split into two classes: 1) those that deal with the alarm handling, e.g. operator interface, design, implementation, maintenance, and 2) those that deal with the alarm contents, e.g. alarm definitions, response procedures, and alarm effectiveness/performance monitoring.
FDA and Alarming
The FDA provides directions for alarm management in two of its guidelines: 1) Computerized Systems in Drug Establishments (2/83) FEBRUARY, 1983, and 2) General Principles of Software Validation; Final Guidance for Industry and FDA Staff, January 11, 2002. The Computerized Systems in Drug Establishments guideline establishes six expectations for alarm management, as listed and classified below:
| Computerized Systems in Drug Establishments Guideline | Requirement Class |
|---|---|
| 1) Documentation of alarm function. The condition that initiates the alarm must be documented. Any interlocks the alarm trigger must be documented. | Alarm Contents |
| 2) Documentation of alarm parameters/thresholds and their maintenance. The alarm set-points must be documented. (Typical values are trigger point and time delay). | Alarm Contents |
| 3) Determination and documentation of alarm response procedure. This expectation goes hand-in-hand with the key expectations for alarm management in the EEMUA standard. | Alarm Contents |
| 4) Definition of how alarms are recorded -in batch records, logs, or automatically by the control system, and maintenance of the history record. It is required to maintain alarm records for all alarms used that indicate out of range conditions. | Alarm Handling |
| 5) Documented design, validation, and maintenance, of the Operator Alarm Interface. (lights, alarm horn, control system Alarm Manager Interface). Maintenance requirements for the interface to ensure it continues to work as validated. E.g. verification of pilot lights. | Alarm Handling |
| 6) The alarm interface must be designed to ensure it captures all alarms and to ensure it provides just in time access to all alarms. The guideline states “Can all alarm conditions be displayed simultaneously or must they be displayed and responded to consecutively? If an employee is monitoring a CRT display covering one phase of the operation will that display alert the employee to an alarm condition at a different phase? If so, how? | Alarm Handling |
483 Observations
The FDA has issued several 483 observations and warning letters whose infractions are traceable to these expectations. For example, the following two statements were extracted from FDA Warning letters: 1) “ There was no data to demonstrate that the chamber alarm would perform as required in the event of a humidity excursion.” 2) “ Alarms for out-of-temperature specification events of incubators, refrigerators and freezers are audible only in the vicinity of the equipment, and there is no system in place to notify employees of these alarms after hours or over the weekend.”
The second guideline, General Principles of Software Validation; Final Guidance for Industry and FDA Staff, January 11, 2002 provides additional expectations for documentation and validation. Among the requirements the guideline specifies the need for two types of documents: User requirements and Software Design specifications. A summary of the areas pertaining to alarm management follows: The user requirements should identify safety related functions including alarms and interlocks logical processing steps, or command sequences. The software design specification should include: Error, alarm, and warning messages. Included with the validation is user site testing to verify Operators can perform the intended functions and respond in an appropriate and timely manner to all alarms, warnings, and error messages.
Alarm Definition and Response Procedure
Alarm management begins with effective definitions of what alarms are required and determination of the alarm response procedures. A discussion on methods for determining the alarming requirements and discussions on effective response process is beyond this review. What follows provides a brief overview.
Alarming requirements should be collected using formal approaches such as Process Hazards Analysis (PHA) and Failure Mode and Effects Analysis (FMEA). Element of a good alarm definition include:
- Documentation of alarm function.
- Documentation of the potential cause(s) that initiate the alarm.
- Documentation of alarm interlocks.
- Documentation of alarm type/setpoint/parameters/thresholds.
- Documentation of escalation procedure.
- Allowable response time
In general, alarm response processes must be tailored to the alarm condition and must be appropriately fitted to the organizational process assets and applicable enterprise environmental factors. The response process must document the step by step actions required to correct the alarm, state the result of inaction, and should include instructions for diagnostic and reporting of root cause condition.
Pharmaceutical Process Alarms Classification and Risk Assessment
Alarms can be categorized into three groups: safety, process controls, and equipment. Within each group, alarm criticality will vary according to function. Alarm prioritization is used to indicate relative criticality between alarms. Alarm prioritization requires expert knowledge about the alarm condition and alarm response. Personnel and Environmental Safety laws provide specific response requirements for safety alarms, and standardize alarm criticality across all industries.
For pharmaceutical process control alarms there are no explicit guidelines for alarm prioritization. The following discussion will illustrate the use of GMP Criticality Assessment and Risk Assessment as tools for alarm prioritization.
Following GAMP recommendations for pharmaceutical facilities and process, all alarms related to pharmaceutical manufacturing should be individually assessed for their criticality to the product. Appropriate assessment requires the use of a suitable risk assessment program.
After consideration of the ISPE guidelines, a model was developed by merging concepts from Instrumentation Management and Hazards Risk Assessment. All alarms directly, or indirectly, involved with the process should be individually assessed for their GMP criticality to the process. The following three categories are suggested:
| GMP Criticality | Description |
|---|---|
| 1 | Product Quality Critical Alarm (Exception Alarm): An alarm whose failure* may have a direct impact on product quality. |
| 2 | Process / System Alarms: An alarm whose failure may affect the process or system performance but does not directly impact product quality. |
| 3 | Non- Critical Alarms: Alarms whose failure has no impact on product quality, systems or the environment. |
GMP Criticality
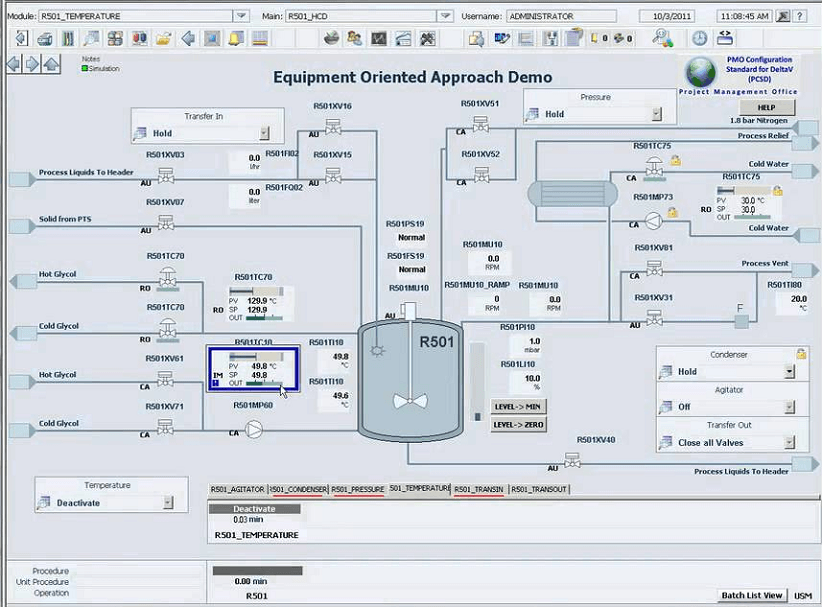
To illustrate GMP Criticality an example alarmed system is shown in Figure 1. The illustration 1 shows a product refrigerator, and temperature controller, whose storage temperature specification is 2 to 7 °C. Drug products must be stored within the specified temperature range.
Quality critical alarms are set at 2 and 7 °C, and performance alarms are placed at 3 and 6 °C, respectively. Clearly, the QA are used to indicate out of tolerance conditions, the performance alarms do not indicate impact in product quality. The key function of the performance alarms would be to warn that the refrigerator controller is not performing and the condition may lead to a critical alarm.

GMP Criticality Assessment results help focus attention on high risk areas and provide adequate basis for determining the scope of alarm validation. Depending on the company risk tolerance level, a company could adopt a policy where only product quality alarms, priority 1 alarms, are validated. A conservative policy will include validation of all Priority 1 and additional Priority 2 alarms that are required to mitigate high risk. Determination of high risk performance alarms is the key to efficient alarm, management.
The use of risk assessment for determining high risk alarms will be demonstrated by examining the product storage freezer warning alarm. The risk assessment model used is a function of two variables: Probability and Severity; and is based on GAMP-5, see Figure 2. In this model the probability that an alarm event will occur is plotted against its severity.
Alarm Description: A high temperature alarm is used to indicate that the product freezer temperature is approaching its maximum temperature. The alarm response procedure requires the operator to place the temperature controller in manual mode and take manual control of the thermostat to lower the temperature to below the warning level.

Alarm Risk Assessment
This alarm has high probability of occurrence and high severity. It has a high probability of occurrence because the condition will occur at some point in time. The alarm is used to prevent impact to product quality. There is a high probability that the temperature would escalate outside the product requirements, if the alarm is left unattended. Hence its severity ranking is High. As indicated with the letter “X” in the Severity vs. Probability plot the alarm has a risk class of 1.
Pharmaceutical companies will be served well by placing appropriate process/system efficiency alarms, to prevent product quality alarms. These alarms should be documented in the Alarm Response Operating Procedures, and validated. Validation is required in order to meet the expectations indicated in the FDA guidelines. The use of process efficiency alarms demonstrates commitment to quality and good business judgment.
Following the product freezer example, it is clear that a Product Temperature critical alarm will result in a discrepancy. And the response process for this QA alarm will be costly. In the worst case product will be lost. In contrast, the entire situation could have been prevented via implementation of performance monitoring warning alarms. Effective implementation of alarm management in FDA regulated pharmaceutical environments requires implementation of suitable process efficiency alarms and alarm handling procedures.
Implementation of exception reporting is a fringe benefit of having appropriate alarm classification and risk assessment. Exception reports are simply a collection of all QA critical alarms generated throughout the manufacturing process.
Each unit operation and the environment must be carefully examined to ensure all necessary QA alarms documented, integrated, tested. Appropriate exception reports can be generated by providing daily QA alarms reports, process cell QA alarm reports. For batch process all QA alarms must be recorded in the batch record, or via batch reports.
In summary two assessments should be performed for alarm classification. 1) GMP criticality assessment, which provides the basis for categorizing alarms based on GMP criticality, and 2) Qualitative Risk assessment, which allows for determining how important the alarm is.
Indicators of poor alarm management in pharmaceutical manufacturing: Proper alarm management is a critical element of pharmaceutical manufacturing. Symptoms of un-compliant, ineffective, alarm management include:
- Undefined alarm prioritization scheme.
- GMP critical alarms are not listed in the SOPs.
- Alarms exist without response procedure, useless alarm messages.
- Existing response procedures are not validated, and/or there is no history that response procedures are followed. Response procedures are incomplete and/or allow for alarms to go unnoticed. Under-monitored alarm system.
- High number of alarm related discrepancies.
- Repetitive alarms: A predictable number of alarms are continuously present in the system and are left unattended because operators learn to ignore them.
Conclusion
Alarm management principles, together with the FDA’s guidelines for risk management and exception reporting, provide the foundation for the implementation of alarm systems in pharmaceutical manufacturing processes. The key principles of Alarm Management include defining the alarms, classifying the alarms, and developing alarm response procedures. The use of GMP classification and risk assessment allows for implementation of exception reporting. Pharmaceutical companies will be well serve by implementing process efficiency alarms systems to prevent production discrepancies.
In addition, when trying to address or satisfy criteria for 21 CFR Part 11 or Annex 11, the definitions implemented as part of an Alarm Management strategy will already be integrated into validation documentation and defined in User Requirements for the system. The use of a risk based approach allows for the end user to satisfy the quality oversight expected from these systems.
Acronyms
GAMP: Good Automated System Manufacturing Practice
ISPE: International Society for Pharmaceutical Engineers
FDA: Food and Drug Administration
SOP: Safety Operating Procedure
Form 483: A form used by the FDA to notify corporations of non-compliance with cGMP regulations